
CO WARTO WIEDZIEĆ ABY ZDROWO ŻYĆ Z ADPKD?
– opowiada nefrolog
ADPKD (Autosomal Dominant Polycystic Kidney Disease) to skrót oznaczający wielotorbielowatość nerek dziedziczoną autosomalnie dominująco. Jest to wrodzona choroba ogólnoustrojowa, prowadząca do nieprawidłowej przebudowy różnych układów i narządów. Najpoważniejsze konsekwencje tych zmian widoczne są w nerkach. W obrębie ich miąższu dochodzi do tworzenia coraz liczniejszych, powiększających się torbieli, które stopniowo zastępują prawidłową tkankę. Nerki stają się duże i przestają prawidłowo działać. Torbiele mogą występować również w innych narządach (wątrobie, trzustce, tarczycy, śledzionie, pęcherzykach nasiennych, pajęczynówce), jednak ich pozanerkowa lokalizacja rzadko wywołuje poważne skutki zdrowotne. Innymi objawami ADPKD mogą być tętniaki naczyń krwionośnych (wewnątrzczaszkowe i aorty), wady zastawki mitralnej serca i przepukliny powłok jamy brzusznej.
ADPKD jest najczęstszą uwarunkowaną genetycznie chorobą obejmującą nerki. Występuje z częstością 1/400 – 1/1000 urodzeń. Dziedziczenie „dominujące“ oznacza, że każde dziecko chorego rodzica ma 50% szans na dziedziczenie tej choroby. Jeśli pacjent z chorobą nie przekaże jej jednemu ze swoich dzieci, wówczas choroba znika z tej rodziny i wnuki nie mogą jej odziedziczyć. Dziedziczenie „autosomalne“ oznacza, że następuje niezależnie od płci i choroba z jednakową częstością dotyczy kobiet jak i mężczyzn.
ADPKD wywołane jest dobrze opisanymi mutacjami w obrębie dwóch genów: PKD1 na chromosomie 16 i PKD2 na chromosomie 4. Jest prawdopodobne, że inne, jeszcze nie znane mutacje genowe mogą być odpowiedzialne za wystąpienie tej choroby. Wśród wszystkich przypadków ADPKD mutacje PKD1 są stwierdzane najczęściej (71-85%). Choroba PKD2 jest łagodniejsza i rozpoznawana w późniejszym okresie życia.
Niektóre osoby z mutacjami genowymi nigdy nie rozwiną objawów ADPKD. Szacuje się, że nawet połowa wszystkich przypadków ADPKD nie jest rozpoznawana z powodu skąpych objawów choroby. Co czwarty pacjent ze świeżo postawionym rozpoznaniem ADPKD jest jedynym członkiem rodziny z jawną manifestacją kliniczną tej choroby, która u pozostałych krewnych przebiega bezobjawowo.
W około 15% przypadków ADPKD występuje u osób z wykluczonym rodzinnym występowaniem choroby (krewni zostali przebadani i nie mają ADPKD). Oznacza to, że pacjent ma nową mutację genetyczną, która nie występowała u żadnego z rodziców.
Zazwyczaj łatwo jest zdiagnozować ADPKD u osób z wywiadem rodzinnym. Podstawowym badaniem przesiewowym jest USG, które ujawnia liczne torbiele w obu nerkach oraz w innych narządach jamy brzusznej. Nerki są zazwyczaj powiększone, choć we wczesnych stadiach choroby mogą mieć normalny rozmiar. ADPKD rozpoznaje się na podstawie uwidocznionej w USG liczby torbieli nerek w zależności od wieku pacjenta i wywiadu. Diagnoza ADPKD zawsze powinna być postawiona lub potwierdzona przez doświadczonego nefrologa.
ADPKD może być trudniejsze do zdiagnozowania u osób bez wywiadu rodzinnego, u których wystąpiła nowa mutacja, lub też członkowie rodziny nie mają dolegliwości albo zmarli przed rozpoznaniem choroby. W takiej sytuacji konieczne jest uwidocznienie w USG przynajmniej 10 torbieli w każdej nerce. Wyniki innych badań obrazowych jamy brzusznej (tomografii komputerowej lub rezonansu magnetycznego) mogą również stanowić podstawę do rozpoznania i powinny być skonsultowane z nefrologiem.
Decyzja o przeprowadzeniu obrazowego badania przesiewowego u osób z grupy ryzyka powinna zostać omówiona z doświadczonym lekarzem. Należy uwzględnić potencjalne zagrożenia (np. psychologiczne) i korzyści z postawienia diagnozy. Zazwyczaj nie zaleca się wykonywania przesiewowego USG u małych dzieci, chyba, że występują wczesne objawy choroby nerek (np. krwinkomocz, białkomocz). Dzieci rodziców z ADPKD powinny być pod stałą opieką nefrologa dziecięcego. W każdym roku powinny mieć wykonany pomiar ciśnienia tętniczego krwi, analizę moczu, obserwację pod kątem objawów zakażenia układu moczowego, kamicy nerkowej i przepuklin.
Badania genetyczne w kierunku identyfikacji mutacji przyczynowej nie są powszechnie wykonywane (mała czułość metody oraz koszty). Decyzja o badaniu w ramach poradnictwa genetycznego zawsze powinna zostać omówiona z doświadczonym nefrologiem. Szczególnym wskazaniem do przeprowadzenia badania genetycznego jest rodzinne dawstwo nerki. Młody dawca nerki, bez widocznych torbieli w badaniu obrazowym, powinien mieć wykluczoną mutację genetyczną przed oddaniem nerki krewnemu z ADPKD. Wynik dodatni będzie przeciwwskazaniem, gdyż nie wiadomo jakie może być tempo rozwoju choroby u osoby po oddaniu nerki. Alternatywnym rozwiązaniem w tej sytuacji jest dawstwo niespokrewnione, czyli pobranie nerki od osoby spoza grupy ryzyka.
Jak już wspomniano, manifestacja kliniczna choroby (tzw. „penetracja genu“) jest bardzo różna i nawet wśród członków jednej rodziny stopień nasilenia i tempo postępu ADPKD bywa skrajnie odmienne. Niewydolność nerek rozwija się zazwyczaj w średnim wieku, lecz nie u wszystkich osiąga stadium schyłkowe. Prawdopodobieństwo wystąpienia konieczności leczenia nerkozastępczego (przeszczepieniem nerki, dializami) szacuje się na mniej niż 2% u osób poniżej czterdziestego roku życia. Wskaźnik ten wzrasta do 50 – 75% dla osób w wieku między 70 a 75 rokiem życia.
Czynniki ryzyka rozwoju niewydolności nerek w przebiegu ADPKD zamieszczono w tabeli numer 1. Najistotniejszym jest duża objętość nerek, oceniona w tomografii komputerowej lub rezonansie magnetycznym. Na wiele czynników ryzyka można aktywnie wpłynąć poprzez zmiany stylu życia. Zaprzestanie palenia papierosów, dieta z ograniczeniem soli, tłuszczów zwierzęcych, aktywność fizyczna, to niektóre ze sposobów pozytywnego wpływu na przebieg ADPKD. Większość osób z ADPKD choruje na nadciśnienie tętnicze (nawet 60-70% pacjentów z jeszcze prawidłową czynnością nerek w wieku 29 lat). Może to być pierwszy objaw tej choroby. Wszystkie osoby zagrożone ADPKD powinny mieć wykonywane regularne pomiary ciśnienia tętniczego. Jeżeli zostanie postawione rozpoznane należy wdrożyć zasady zdrowego stylu życia oraz rozpocząć leczenie farmakologiczne pod nadzorem nefrologa.

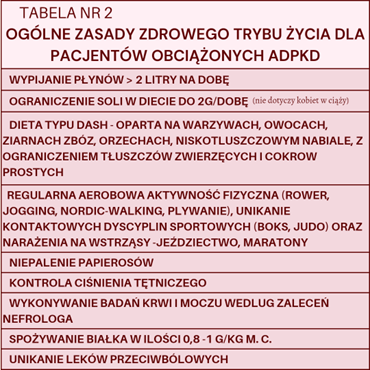
Zasady zdrowego stylu życia dla osób z ADPKD przedstawiono w tabeli numer 2. W szczególności należy zwrócić uwagę na brak konieczności ograniczenia białka w diecie oraz aktywności fizycznej. ADPKD nie stanowi medycznego przeciwwskazania do prokreacji. Ciążę trzeba jednak zaplanować, gdyż mogą być konieczne wcześniejsze zmiany w lekach. Podczas ciąży należy odbywać regularne wizyty u nefrologa.
Obecność krwi w moczu (hematuria), występuje u 35-50% osób z ADPKD i może być pierwszym objawem choroby. Pojedynczych krwinek czerwonych nie widać gołym okiem, ujawnia je laboratoryjna analiza moczu. Epizody hematurii bywają częste. Przyczyną jest krwawienie do torbieli komunikującej się z drogami moczowymi. Leczenie obejmuje odpoczynek w łóżku i zwiększanie ilości przyjmowanych płynów, aż do ustąpienia krwawienia (2-7 dni). Jeśli to nie nastąpi – konieczny jest kontakt z lekarzem. Krwawienie do torbieli, która nie komunikuje się z układem moczowym, może objawić się tylko bólem i gorączką, bez krwiomoczu.
Podstawowym objawem powikłania w postaci bakteryjnego zakażenia torbieli jest gorączka często z bólem w okolicy lędźwiowej. Leczenie odpowiednio dobranym antybiotykiem, penetrującym do torbieli, powinien prowadzić nefrolog. Antybiotykoterapia jest zazwyczaj dłuższa, niż w przypadku zakażenia u osoby bez ADPKD. Niektóre osoby z wysoką gorączką lub ciężkimi objawami wymagają hospitalizacji i dożylnego podawania płynów oraz leków.
U co czwartej osoby z ADPKD rozpoznaje się kamicę dróg moczowych, która może powodować ból, hematurię lub blokadę przepływu moczu. Leczenie kamicy moczowej oraz usuwanie złogów blokujących przepływ moczu jest niezwykle ważne u pacjentów z ADPKD. Jeśli blokada nie zostanie skutecznie usunięta może nastąpić szybka i trwała utrata funkcji nerki.
Szacuje się, że 25% osób z ADPKD może mieć wadę serca, najczęściej bezobjawową, i tylko w wyjątkowych przypadkach konieczna jest operacja kardiochirurgiczna.
Najpoważniejszym z powikłań w przebiegu ADPKD jest pęknięcie tętniaka tętnicy podstawy mózgu. Pierwszym objawem jest nagły, ostry ból głowy z towarzyszącymi wymiotami. Jest to stan zagrożenia życia i należy niezwłocznie zgłosić się po pomoc medyczną. Tętniak to wybrzuszenie w ścianie naczynia krwionośnego wywołane odcinkowym osłabieniem w jej budowie. Ryzyko pęknięcia tętniaka mózgu wzrasta, gdy jego średnica przekracza 10 mm. W porównaniu z populacją ogólną, tętniaki mózgu u chorych z ADPKD występują 5 razy częściej. Nie ma dokładnie określonych wskazań do wykonywania badań przesiewowych w kierunku wykrycia a następnie profilaktycznego leczenia tętniaka u osób z ADPKD. Warto je wykonać przed planowanym leczeniem przeciwkrzepliwym, przed transplantacją nerki, oraz gdy w rodzinie występują osoby z tętniakiem, po przebytym udarze mózgu, ze skargami na nietypowe bóle głowy.
Dużo łagodniejszym powikłaniem są przepukliny w obrębie powłok jamy brzusznej (pachwinowa, pępkowa), obecne u blisko połowy osób z ADPKD.
Często u pacjentów z ADPKD występują torbiele w miąższu wątroby, widoczne już przed 30 rokiem życia. Torbiele wątroby najczęściej nie dają żadnych dolegliwości i nie upośledzają czynności tego narządu. Duże i mnogie torbiele mogą wywoływać uczucie pełności jamy brzusznej, brak apetytu, duszność oraz tępy ból w prawym podżebrzu.
Jeśli dojdzie do schyłkowej niewydolności nerek w przebiegu ADPKD należy rozpocząć leczenie nerkozastępcze (zastąpić czynność niedziałających i zniszczonych nerek). Najlepszą metodą jest przeszczepienie nerki. Optymalnie, aby odbyło się w trybie przeddializacyjnym, od dawcy żywego. Konieczne jest jak najszybsze wykonanie badań kwalifikujących do umieszczenia na Krajowej Liście Oczekujących na przeszczepienie nerki (KLO). Przed transplantacją chirurg może zalecić usunięcie bardzo powiększonej nerki własnej, chociaż współcześnie zaleca się aby minimalizować wskazania do tej operacji. Poza brakiem przestrzeni na nowy narząd, nefrektomię podejmuje się w przypadku silnego bólu, krwiomoczu wymagającego przetoczeń krwi oraz podejrzenia nowotworu torbieli. Coraz częściej taki zabieg wykonywany jest metodą laparoskopową. Przeżywalność nerki przeszczepionej u chorych z ADPKZ jest lepsza w porównaniu do pacjentów z inną przyczyną niewydolności nerek. W trakcie badań i oczekiwania na przeszczepienie nerki, lub w przypadku istnienia trwałych lub czasowych przeciwwskazań do transplantacji, konieczne będzie planowe rozpoczęcie dializoterapii.
Pacjenci z ADPKD powinni jak najwięcej wiedzieć o swojej chorobie, o zagrożeniach z nią związanych, o zasadach jej monitorowania i leczenia. Wiedza ta pozwala kontrolować chorobę, cieszyć się pełnią życia oraz osiągać sukcesy w sferze rodzinnej i zawodowej.
Dr n. med. Marta Serwańska-Świętek, specjalista chorób wewnętrznych, nefrologii i transplantologii klinicznej. Z-ca Dyrektora Medycznego DaVita Polska.